
16S rRNA sequencing is a powerful technique used to identify and classify bacteria and archaea based on their genetic material. It targets the 16S ribosomal RNA gene, which is present in all prokaryotes and contains both conserved and variable regions.
Common methodologies
Sanger Sequencing
Read length is typically ~700–1,000 bp, covering the full-length 16S rRNA gene. This method is high-quality and low-throughput, making it useful for identifying single isolates or pure cultures. It is accurate and well-established but not practical for complex or mixed microbial communities. Commonly used in pure culture applications rather than community analysis (e.g., activated sludge).
Next-Generation Sequencing
Platforms such as Illumina (most common) and Ion Torrent produce reads of 150–300 bp, with paired-end reads reaching up to 600 bp. This approach is used in microbiome studies and environmental samples. It is high throughput and cost-effective, though shorter reads can limit taxonomic resolution. Common target regions for Illumina NGS include V1–V3, V3–V4 (most popular), V4, and V4–V5.
Third-Generation (Long-Read) Sequencing
Platforms like PacBio SMRT and Oxford Nanopore produce full-length 16S reads (~1,500 bp) for improved taxonomic resolution down to the species level. These methods provide long reads and full-gene coverage but have higher error rates (which are improving) and are generally costlier than NGS.
Practical takeaways
Not all DNA testing is the same. Different methodologies often produce different results, so it is important to know what method is being used when comparing results. Separate testing is commonly used for bacteria vs. archaea. For example, in anaerobic systems methanogens are archaea and testing for both is recommended.
What is actually measured in rRNA testing?
A useful analogy is that “bugs” have varying numbers of “fingers,” with rRNA acting as a way of counting fingers. DNA read percentage is therefore not always a strong correlation to actual organism abundance.
Based on hundreds of DNA/microscopy split samples, it appears common for as low as 2–3% of DNA reads from a genus such as Microthrix to correlate with abundant filaments. In contrast, genera such as Thauera (commonly with zoogloea morphology) may often register around 20% with corresponding zoogloea bacteria type abundance at "common" to "very common" using the Jenkins 1–6 ranking scale.
In some but not all instances, trends may be useful. However, DNA read percentage and microorganism abundance are not an apples to apples comparison based on the variability of rRNA per given microbe.
Other potential factors include that faster-growing microbes are more likely to have higher DNA read percentages, microbes with higher polysaccharide tend to have higher DNA read percentages, and slow-growing filaments such as those within the Chloroflexi phylum may produce low DNA read percentages despite high abundance under the microscope. For example, it is common for around 1% Kouleothrix genus reads to correlate with very common–abundant type 1851 filaments.
Correlation with genus reads is likely more representative as a “who’s there” rather than a detailed expression of abundance. Microscopy is a useful tool for morphotype (what something looks like) abundance, and DNA can complement this by confirming “who’s there” at various taxonomic levels. Morphotype/cause correlations have generally held up strongly for over 40 years of operational practice, especially when considering the surrounding microbes viewed within a sample.
DNA: functional groups and morphology
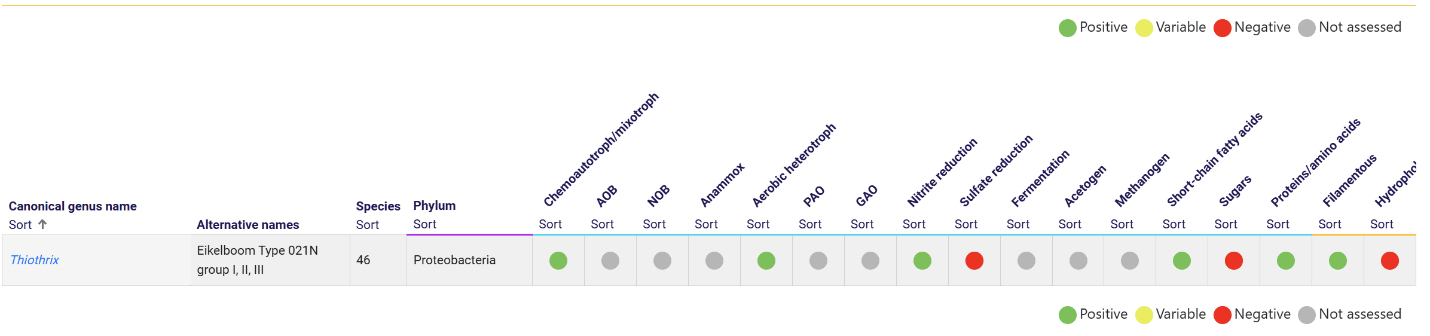
When referencing a genus using the MiDAS field guide, there are highlighted regions for positive, variable, negative and unknown functionalities. Many genera have variable potential functionalities, so just because a microbe is capable of doing something (often at bench scale vs. in situ) does not confirm the role it is actually serving in biological wastewater treatment.
Most genera capable of filamentous form are labeled “variable,” as they may or may not have filamentous morphology under the microscope. Many have overlapping potential functionality, such as PAOs that may also be GAOs (e.g., Ca. Accumulibacter). Complexities are further amplified because many genera are capable of oxidation and/or fermentation reactions, and even in processes such as enhanced biological phosphorus removal, many of these genera may also be capable of denitrification.
DNA is an emerging and growing field of research, and some genera have more available recognized information than others at the time of this writing. In practice, it is common that varying percentages of reads do not reach low taxonomic levels (this will be discussed in further detail later within this article).
What’s in a name?
There is not a universally accepted database for microbe taxonomic classification. Personally, the MiDAS field guide appears to be the most practical resource; however, other credible and recognized sources include NCBI, LPSN, GTDB, SILVA, RDP, EzBioCloud and BacDive. Taxonomy, genus names, and more may differ depending on what database is used, so this is important context.
On the microscopy side, there is generally broad potential genetic diversity within various recognized morphological descriptions. For example, there are 46 recognized species of Thiothrix, meaning even within the genus level, they are not always the same. There are also many closely related microbes that are capable of displaying Thiothrix morphology but do not actually belong to the Thiothrix genus.
Basic information on taxonomy

This example portion of a DNA report (courtesy of Aster Bio) using the Oxford Nanopore platform is based predominantly on the MiDAS database, along with added information from other credible sources. Reads in this instance cover kingdom, phylum, class, order, family and genus, and report as low on the chart as accurately determined by the program. Blank spaces indicate that the margin of error was not within the designated range for confident classification at a particular taxonomic level.
Practical takeaways
DNA is an evolving field, and as more knowledge is obtained, occasional changes to classifications will occur. As technology improves, the criteria for genus classification in many instances is narrowing. It is important to consider not only genus reads but also higher taxonomic orders, where functionality may capture a wider range of the big-picture conditions.
Many DNA reports on functional groups may be based on genus-level reads, but there may be varying amounts of reads that do not reach the genus level. Some phyla show general functional and morphological consistency (e.g., Chloroflexi as slower growing filamentous bacteria, many capable of storing substrate under anaerobic conditions), while others show significantly more diversity (e.g., Nitrosomonas and Thiothrix belong to the same phylum).
Benefits and limitations of DNA testing
DNA testing is repeatable and non-subjective, provided there is known information about testing methods and databases used. Databases are growing at a high rate, with more information being learned about microbes at bench scale, in pure culture and in situ. While read percentage is not always a strong and reliable comparison to microbe abundance as viewed under the microscope, trends are effective in many instances depending on objectives. Technology is advancing, and pricing and turnaround time for DNA testing are becoming more practical.
However, DNA testing does not provide reliable context for conditions such as floc structure characteristics (firm vs. diffuse floc), dispersed growth abundance or bacterial health/viability. Higher life form organisms (protozoa, metazoa) and fungi require different methodologies than community analysis for bacteria. There is variable functionality and morphology among many genera. DNA is an excellent tool for establishing if a microbe is present, but if morphology and functionality are variable, other tools such as microscopy and in-house analytical testing may be needed for context. For example, Acinetobacter genus is commonly correlated with type 1863 filaments but does not always display that morphology under the microscope.
Tools such as microscopy, in-house analytical testing, DNA testing, and operator intuition are all relevant and useful. Gathering information from multiple sources gives the highest levels of baseline data for troubleshooting and operational process control. Tools and methodologies are only as good as the quality with which they are obtained, and knowing the context and potential variables is key for practical application.
About the author: Ryan Hennessy is the principal scientist at Ryan Hennessy Wastewater Microbiology. He was trained and mentored by Dr. Michael Richard for over 10 years in wastewater microbiology, and serves as a microbiology services consultant. Hennessy is a licensed wastewater treatment and municipal waterworks operator in the state of Wisconsin and fills in as needed for operations at several facilities. He can be reached at ryan@rhwastewatermicrobiology.com. Hennessy's new book Wastewater Microbiology: Filamentous Bacteria Morphotype Identification Techniques, and Process Control Troubleshooting Strategies is now available on Amazon.














